The liver is a vital organ located in the upper right side of the abdomen. In an adult, it weighs about 1.2 kg. It is one of the most important organs in the body and keeps us healthy by performing many essential functions.
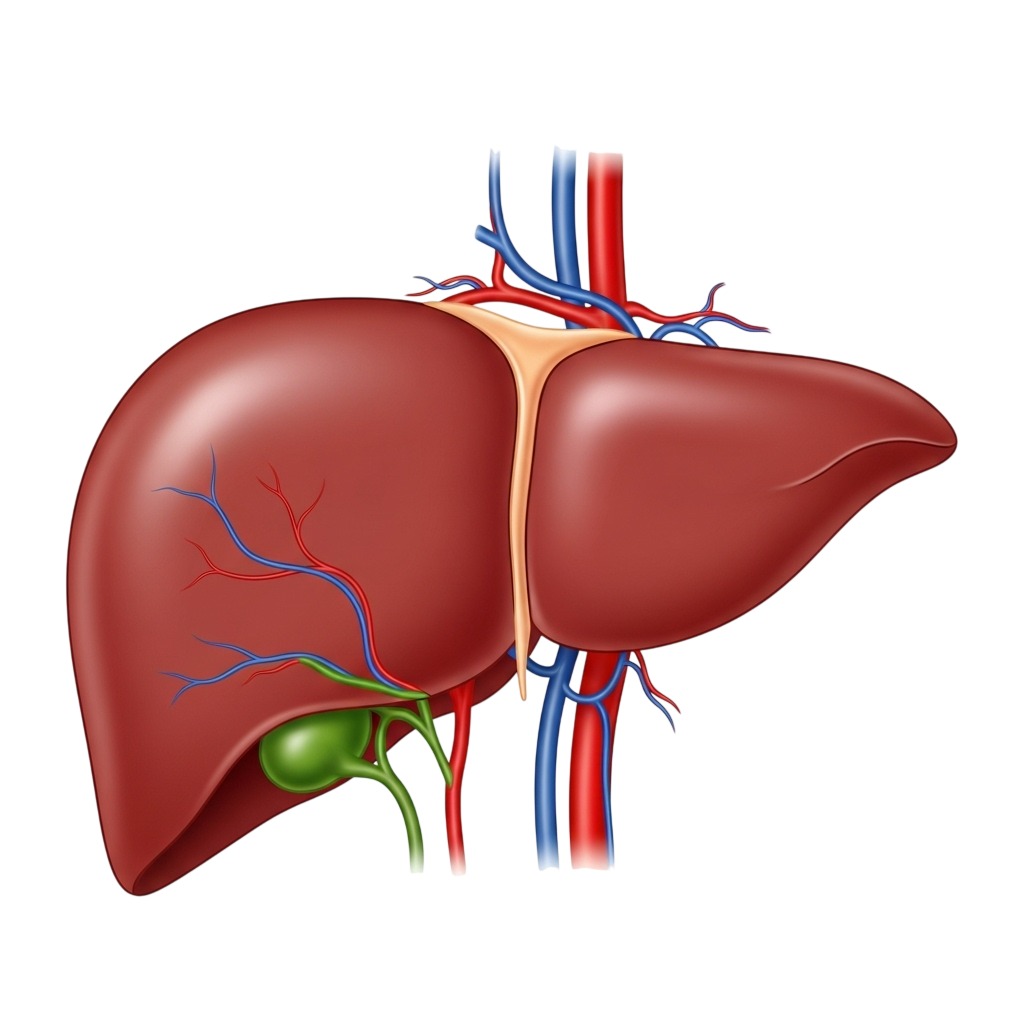
After you eat, blood from the stomach and intestines passes through the liver.
The liver
then:
These are needed for growth, development, and daily functioning.
The liver makes bile, a fluid that helps digest fats and absorb fat-soluble vitamins.
It produces special proteins that prevent excessive bleeding when you get hurt.
The liver acts as the body’s natural filter, removing drugs, toxins, and harmful substances from the bloodstream.
The liver plays a role in recognizing and fighting infections, helping keep the body safe.
Liver disease can occur due to many different reasons, including (Understanding the cause helps in early diagnosis and effective treatment.):
| CHILDREN | ADULTS |
|---|---|

In children, liver diseases are usually:
Common examples include biliary atresia, metabolic liver diseases, and certain genetic disorders. |

In adults, liver diseases are mostly acquired later in life, commonly due to:
|
Children with liver disease need a dedicated pediatric hepatology and transplant team, as their conditions progress differently, and treatment must be tailored to their growth and development.
Jaundice is common in newborns, but when it lasts beyond two weeks in full-term babies or three weeks in preterm babies, it is considered prolonged or pathological jaundice. This type of jaundice needs prompt medical evaluation, as it may indicate an underlying liver, metabolic, or anatomical problem.
While blood group incompatibility (such as Rhesus or ABO incompatibility) is a well-known cause, many other conditions can lead to jaundice during the neonatal period.
1. Anatomical Liver and Bile Duct Disorders
Biliary atresia — a serious condition where bile ducts are blocked or absent
Liver cysts and other structural abnormalities
These require early diagnosis because delayed treatment can permanently damage the liver.
2. Hereditary and Genetic Conditions
Progressive Familial Intrahepatic Cholestasis (PFIC)
Alagille syndrome
These disorders affect bile flow and can present with jaundice very early in life.
3. Metabolic Liver Disorders
Galactosemia
Tyrosinemia
These are rare but serious conditions where the baby cannot process certain nutrients, leading to liver injury and jaundice.
Timely identification of the cause of prolonged jaundice is essential to:
Prevent liver damage
Start appropriate treatment early
Reduce the risk of complications
Protect the baby’s long-term health and development
If your newborn has jaundice that does not improve after two to three weeks, seek medical care immediately. Early action can make all the difference.
Biliary atresia is a serious liver and bile duct disease in newborns where the bile ducts—the channels that carry bile from the liver to the intestine—are blocked, narrowed, or completely absent. Because bile cannot flow properly, it builds up inside the liver and causes jaundice, liver inflammation, and damage.
Early detection is essential because timely surgery greatly improves outcomes. The best results occur when the corrective surgery, called portoenterostomy (Kasai procedure), is performed within the first 2–3 months of life.
Success Rates of Surgery
More than 50% of infants operated within 2–3 months of age can successfully clear their jaundice
These children have over an 80% chance of growing well and reaching adolescence without needing a liver transplant
Early diagnosis is the key factor that influences long-term success
Parents should seek urgent medical attention if their baby has:
1. Pale or Clay-Colored Stools
Biliary atresia often causes pale, whitish, or clay-colored stools, because bile is not reaching the
intestine.
(Refer to a stool colour chart to check your baby’s stool.)
2. Dark Yellow or Brownish Urine
When pale stools occur along with dark urine, this strongly suggests a blockage in bile drainage.
3. Jaundice Lasting More Than 2 Weeks
Persistent jaundice beyond 14 days in a full-term baby is a red flag for neonatal cholestasis, and biliary atresia must be ruled out immediately.
If your newborn has pale stools, dark urine, or jaundice that does not improve, consult your pediatrician
or pediatric hepatologist right away.
Early evaluation can make the difference between successful surgical treatment and the need for a liver
transplant later in childhood.
Progressive Familial Intrahepatic Cholestasis (PFIC) is a group of genetic liver diseases in children where bile cannot flow properly from the liver. This condition is relatively common in India and is one of the important causes of chronic cholestasis in infants and young children.
Children with PFIC often show signs early in life. Common symptoms include:
Mild to severe jaundice
Severe itching (pruritus) due to high bile acids
Low GGT levels on blood tests(in few subtypes)
Very high serum bile acids
Poor weight gain or failure to thrive
Chronic diarrhea in some types due to intestinal involvement
There are more than ten PFIC types, but PFIC Type 1, Type 2, and Type 3 are the most common in Indian children.
1. Medical Management
Milder forms of PFIC may respond to medications that reduce itching or improve bile flow. These medicines can help control symptoms and improve day-to-day comfort.
2. Biliary Diversion Surgery
If the itching is severe or bile acids remain very high, a biliary diversion procedure may be advised.
This surgery reroutes bile either:
Externally (to a stoma), or
Internally (within the intestine)
This helps reduce the total bile acid levels, easing itching, improving jaundice, and enhancing the child’s quality of life.
3. Liver Transplantation
When PFIC progresses to cirrhosis or liver failure, a liver transplant becomes the only long-term cure.
A successful transplant not only replaces the damaged liver but also corrects the underlying metabolic
problem.
Children with PFIC have a higher risk of developing liver cancer (hepatocellular carcinoma). Because of
this, they require regular follow-up, blood tests, and ultrasound scans throughout their life.
Early diagnosis, timely surgery, and appropriate treatment significantly improve long-term outcomes.
Acute liver failure (ALF) is a sudden and severe loss of liver function in a previously healthy child. It develops rapidly—often within days—and leads to jaundice, impaired blood clotting, and failure of the liver to perform its essential functions.
ALF can be triggered by several conditions, including:
Infections (e.g., viral hepatitis like Hepatitis A or Hepatitis E)
Metabolic liver diseases
Autoimmune liver disorders
Genetic conditions, such as Wilson’s disease
Toxins or drugs
However, in 30–40% of children, even after extensive tests, no clear cause is found. This is known as indeterminate or seronegative acute liver failure.
Acute liver failure progresses very quickly. Without timely treatment, it can become life-threatening within days. Early identification and urgent medical intervention can save a child’s life.
Some conditions—such as Wilson’s disease and autoimmune hepatitis—often do not respond well to medical treatment and may require liver transplantation.
Certain metabolic liver diseases, on the other hand, may improve with specific dietary modifications, making correct diagnosis extremely important.
Determining the exact cause of acute liver failure is essential because it guides the type of treatment and helps decide whether the child needs:
Standard liver transplantation, or
Auxiliary liver transplantation
An auxiliary liver transplant involves placing a partial donor liver next to the child’s own liver, rather than replacing it entirely.
This approach is particularly useful for acute liver failure caused by infections such as Hepatitis A or Hepatitis E, where the native liver has the potential to recover fully.
A portion of donor liver is transplanted
The child’s own liver begins to regenerate once the acute phase settles
Immunosuppressive medicines can be gradually stopped
Over time, the transplanted segment shrinks and becomes inactive
The child can eventually live with their completely recovered native liver
Alagille syndrome is a genetic, multisystem disorder that affects several parts of the body, especially the liver, heart, eyes, bones, and facial features.
1. Characteristic Facial Appearance
Children with Alagille syndrome often have a distinctive look, including:
A triangular-shaped face
Broad forehead
Pointed chin
Deep-set eyes
These features become more noticeable as the child grows.
2. Cholestatic Liver Disease
The liver is commonly affected due to reduced or malformed bile ducts, leading to:
Persistent jaundice
Itching (pruritus)
Poor growth
Fat-soluble vitamin deficiencies
Liver disease can range from mild cholestasis to severe liver failure.
3. Heart Abnormalities
The most frequent heart defect is:
Peripheral pulmonary stenosis
Some children may have additional congenital heart issues that need cardiology evaluation.
4. Eye Findings
A classic eye feature in Alagille syndrome is:
Posterior embryotoxon
This is harmless by itself but helps in diagnosis.
5. Vertebral (Spine) Changes
Many children show:
Butterfly vertebrae
This is usually seen on X-ray and rarely causes symptoms.
The severity of Alagille syndrome varies widely:
Mild Cases
Itching and cholestasis can often be managed with medications, nutritional support, and fat-soluble vitamin supplementation.
Severe Liver Disease
Children with progressive liver damage, poor growth, intractable itching, or liver failure
may require liver transplantation.
Transplant outcomes for Alagille syndrome are generally excellent.
Caroli’s disease is a rare congenital liver disorder in which the bile ducts inside the liver become abnormally enlarged and form multiple cyst-like dilations. These abnormal bile ducts can trap bile, making the liver prone to recurrent infections (cholangitis), which may lead to serious complications including sepsis.
Caroli’s disease can occur on its own, but in some newborns it appears along with autosomal recessive polycystic kidney disease (ARPKD).
In these infants:
The bile ducts may show extensive cystic changes
However, kidney failure often becomes the main clinical problem
Both liver and kidney involvement influence long-term outcomes
Diagnosis usually involves imaging studies:
1. Ultrasound (USG Liver)
Often sufficient to suggest Caroli’s disease
Shows characteristic cystic dilations within the liver
2. Cholangiography (MRCP / ERCP)
Provides definitive confirmation
Clearly outlines the shape and connection of the dilated bile ducts
The prognosis in Caroli’s disease depends on:
Liver-related complications
Recurrent bile duct infections (cholangitis)
Fibrosis and scarring of the liver
Portal hypertension
Kidney involvement
Severity of polycystic kidney disease
Degree of renal dysfunction
Children with frequent infections or significant liver damage may eventually require liver transplantation, and those with severe ARPKD may require combined liver–kidney transplant.
Non-Alcoholic Fatty Liver Disease (NAFLD) is becoming one of the most common liver problems in children, especially in India. It occurs when excess fat builds up in the liver of children who do not consume alcohol. NAFLD is strongly linked to the rising rates of childhood obesity, unhealthy diets, and reduced physical activity.
With rapid lifestyle changes, many children today consume high-calorie, fast foods such as fried snacks, burgers, pizzas, and sugary drinks. Combined with reduced outdoor play and physical activity, this leads to weight gain, which increases the risk of developing fatty liver disease.
Childhood obesity is often underestimated, but it can cause serious health issues early in life, including fatty liver, diabetes, and high cholesterol.
3-10% of children with normal weight may have NAFLD
50–70% of obese children have fatty liver disease
This makes NAFLD one of the leading causes of chronic liver disease in children.
Most children have no symptoms, and NAFLD is often detected during routine ultrasound screening. Some children may experience:
Fatigue
Abdominal discomfort
Mild overweight-related symptoms
As the disease progresses, inflammation and scarring (NASH and fibrosis) can develop.
There is no specific medicine for pediatric NAFLD.The most effective treatment is lifestyle modification, which includes:
Regular physical activity (at least 1 hour/day)
Healthy, balanced diet with fewer fried and processed foods
Reducing sugary drinks and junk foods
Maintaining a healthy weight
These steps can significantly improve liver health and reverse early disease.
If left untreated, NAFLD can slowly worsen and lead to:
Liver inflammation (NASH)
Liver fibrosis
Cirrhosis
Increased risk of liver failure
In severe, untreated cases, NAFLD can progress to the point where a liver transplant may be needed in adulthood.
Jaundice is common in newborns, but when it lasts beyond two weeks in full-term babies or three weeks in preterm babies, it is considered prolonged or pathological jaundice. This type of jaundice needs prompt medical evaluation, as it may indicate an underlying liver, metabolic, or anatomical problem.
While blood group incompatibility (such as Rhesus or ABO incompatibility) is a well-known cause, many other conditions can lead to jaundice during the neonatal period.
1. Anatomical Liver and Bile Duct Disorders
Biliary atresia — a serious condition where bile ducts are blocked or absent
Liver cysts and other structural abnormalities
These require early diagnosis because delayed treatment can permanently damage the liver.
2. Hereditary and Genetic Conditions
Progressive Familial Intrahepatic Cholestasis (PFIC)
Alagille syndrome
These disorders affect bile flow and can present with jaundice very early in life.
3. Metabolic Liver Disorders
Galactosemia
Tyrosinemia
These are rare but serious conditions where the baby cannot process certain nutrients, leading to liver injury and jaundice.
Timely identification of the cause of prolonged jaundice is essential to:
Prevent liver damage
Start appropriate treatment early
Reduce the risk of complications
Protect the baby’s long-term health and development
If your newborn has jaundice that does not improve after two to three weeks, seek medical care immediately. Early action can make all the difference.
Biliary atresia is a serious liver and bile duct disease in newborns where the bile ducts—the channels that carry bile from the liver to the intestine—are blocked, narrowed, or completely absent. Because bile cannot flow properly, it builds up inside the liver and causes jaundice, liver inflammation, and damage.
Early detection is essential because timely surgery greatly improves outcomes. The best results occur when the corrective surgery, called portoenterostomy (Kasai procedure), is performed within the first 2–3 months of life.
Success Rates of Surgery
More than 50% of infants operated within 2–3 months of age can successfully clear their jaundice
These children have over an 80% chance of growing well and reaching adolescence without needing a liver transplant
Early diagnosis is the key factor that influences long-term success
Parents should seek urgent medical attention if their baby has:
1. Pale or Clay-Colored Stools
Biliary atresia often causes pale, whitish, or clay-colored stools, because bile is not reaching the
intestine.
(Refer to a stool colour chart to check your baby’s stool.)
2. Dark Yellow or Brownish Urine
When pale stools occur along with dark urine, this strongly suggests a blockage in bile drainage.
3. Jaundice Lasting More Than 2 Weeks
Persistent jaundice beyond 14 days in a full-term baby is a red flag for neonatal cholestasis, and biliary atresia must be ruled out immediately.
If your newborn has pale stools, dark urine, or jaundice that does not improve, consult your pediatrician
or pediatric hepatologist right away.
Early evaluation can make the difference between successful surgical treatment and the need for a liver
transplant later in childhood.
Progressive Familial Intrahepatic Cholestasis (PFIC) is a group of genetic liver diseases in children where bile cannot flow properly from the liver. This condition is relatively common in India and is one of the important causes of chronic cholestasis in infants and young children.
Children with PFIC often show signs early in life. Common symptoms include:
Mild to severe jaundice
Severe itching (pruritus) due to high bile acids
Low GGT levels on blood tests(in few subtypes)
Very high serum bile acids
Poor weight gain or failure to thrive
Chronic diarrhea in some types due to intestinal involvement
There are more than ten PFIC types, but PFIC Type 1, Type 2, and Type 3 are the most common in Indian children.
1. Medical Management
Milder forms of PFIC may respond to medications that reduce itching or improve bile flow. These medicines can help control symptoms and improve day-to-day comfort.
2. Biliary Diversion Surgery
If the itching is severe or bile acids remain very high, a biliary diversion procedure may be advised.
This surgery reroutes bile either:
Externally (to a stoma), or
Internally (within the intestine)
This helps reduce the total bile acid levels, easing itching, improving jaundice, and enhancing the child’s quality of life.
3. Liver Transplantation
When PFIC progresses to cirrhosis or liver failure, a liver transplant becomes the only long-term cure.
A successful transplant not only replaces the damaged liver but also corrects the underlying metabolic
problem.
Children with PFIC have a higher risk of developing liver cancer (hepatocellular carcinoma). Because of
this, they require regular follow-up, blood tests, and ultrasound scans throughout their life.
Early diagnosis, timely surgery, and appropriate treatment significantly improve long-term outcomes.
Acute liver failure (ALF) is a sudden and severe loss of liver function in a previously healthy child. It develops rapidly—often within days—and leads to jaundice, impaired blood clotting, and failure of the liver to perform its essential functions.
ALF can be triggered by several conditions, including:
Infections (e.g., viral hepatitis like Hepatitis A or Hepatitis E)
Metabolic liver diseases
Autoimmune liver disorders
Genetic conditions, such as Wilson’s disease
Toxins or drugs
However, in 30–40% of children, even after extensive tests, no clear cause is found. This is known as indeterminate or seronegative acute liver failure.
Acute liver failure progresses very quickly. Without timely treatment, it can become life-threatening within days. Early identification and urgent medical intervention can save a child’s life.
Some conditions—such as Wilson’s disease and autoimmune hepatitis—often do not respond well to medical treatment and may require liver transplantation.
Certain metabolic liver diseases, on the other hand, may improve with specific dietary modifications, making correct diagnosis extremely important.
Determining the exact cause of acute liver failure is essential because it guides the type of treatment and helps decide whether the child needs:
Standard liver transplantation, or
Auxiliary liver transplantation
An auxiliary liver transplant involves placing a partial donor liver next to the child’s own liver, rather than replacing it entirely.
This approach is particularly useful for acute liver failure caused by infections such as Hepatitis A or Hepatitis E, where the native liver has the potential to recover fully.
A portion of donor liver is transplanted
The child’s own liver begins to regenerate once the acute phase settles
Immunosuppressive medicines can be gradually stopped
Over time, the transplanted segment shrinks and becomes inactive
The child can eventually live with their completely recovered native liver
Alagille syndrome is a genetic, multisystem disorder that affects several parts of the body, especially the liver, heart, eyes, bones, and facial features.
1. Characteristic Facial Appearance
Children with Alagille syndrome often have a distinctive look, including:
A triangular-shaped face
Broad forehead
Pointed chin
Deep-set eyes
These features become more noticeable as the child grows.
2. Cholestatic Liver Disease
The liver is commonly affected due to reduced or malformed bile ducts, leading to:
Persistent jaundice
Itching (pruritus)
Poor growth
Fat-soluble vitamin deficiencies
Liver disease can range from mild cholestasis to severe liver failure.
3. Heart Abnormalities
The most frequent heart defect is:
Peripheral pulmonary stenosis
Some children may have additional congenital heart issues that need cardiology evaluation.
4. Eye Findings
A classic eye feature in Alagille syndrome is:
Posterior embryotoxon
This is harmless by itself but helps in diagnosis.
5. Vertebral (Spine) Changes
Many children show:
Butterfly vertebrae
This is usually seen on X-ray and rarely causes symptoms.
The severity of Alagille syndrome varies widely:
Mild Cases
Itching and cholestasis can often be managed with medications, nutritional support, and fat-soluble vitamin supplementation.
Severe Liver Disease
Children with progressive liver damage, poor growth, intractable itching, or liver failure
may require liver transplantation.
Transplant outcomes for Alagille syndrome are generally excellent.
Caroli’s disease is a rare congenital liver disorder in which the bile ducts inside the liver become abnormally enlarged and form multiple cyst-like dilations. These abnormal bile ducts can trap bile, making the liver prone to recurrent infections (cholangitis), which may lead to serious complications including sepsis.
Caroli’s disease can occur on its own, but in some newborns it appears along with autosomal recessive polycystic kidney disease (ARPKD).
In these infants:
The bile ducts may show extensive cystic changes
However, kidney failure often becomes the main clinical problem
Both liver and kidney involvement influence long-term outcomes
Diagnosis usually involves imaging studies:
1. Ultrasound (USG Liver)
Often sufficient to suggest Caroli’s disease
Shows characteristic cystic dilations within the liver
2. Cholangiography (MRCP / ERCP)
Provides definitive confirmation
Clearly outlines the shape and connection of the dilated bile ducts
The prognosis in Caroli’s disease depends on:
Liver-related complications
Recurrent bile duct infections (cholangitis)
Fibrosis and scarring of the liver
Portal hypertension
Kidney involvement
Severity of polycystic kidney disease
Degree of renal dysfunction
Children with frequent infections or significant liver damage may eventually require liver transplantation, and those with severe ARPKD may require combined liver–kidney transplant.
Non-Alcoholic Fatty Liver Disease (NAFLD) is becoming one of the most common liver problems in children, especially in India. It occurs when excess fat builds up in the liver of children who do not consume alcohol. NAFLD is strongly linked to the rising rates of childhood obesity, unhealthy diets, and reduced physical activity.
With rapid lifestyle changes, many children today consume high-calorie, fast foods such as fried snacks, burgers, pizzas, and sugary drinks. Combined with reduced outdoor play and physical activity, this leads to weight gain, which increases the risk of developing fatty liver disease.
Childhood obesity is often underestimated, but it can cause serious health issues early in life, including fatty liver, diabetes, and high cholesterol.
3-10% of children with normal weight may have NAFLD
50–70% of obese children have fatty liver disease
This makes NAFLD one of the leading causes of chronic liver disease in children.
Most children have no symptoms, and NAFLD is often detected during routine ultrasound screening. Some children may experience:
Fatigue
Abdominal discomfort
Mild overweight-related symptoms
As the disease progresses, inflammation and scarring (NASH and fibrosis) can develop.
There is no specific medicine for pediatric NAFLD.The most effective treatment is lifestyle modification, which includes:
Regular physical activity (at least 1 hour/day)
Healthy, balanced diet with fewer fried and processed foods
Reducing sugary drinks and junk foods
Maintaining a healthy weight
These steps can significantly improve liver health and reverse early disease.
If left untreated, NAFLD can slowly worsen and lead to:
Liver inflammation (NASH)
Liver fibrosis
Cirrhosis
Increased risk of liver failure
In severe, untreated cases, NAFLD can progress to the point where a liver transplant may be needed in adulthood.







